David A Knowles
Core Member, New York Genome Center
Assistant Professor, Computer Science, Columbia University
This is my old personal website. The website for my lab at the New York Genome Center and Columbia University is here.
I was previously a postdoc at Stanford with Sylvia Plevritis (Center for Computational Systems Biology/Radiology) and Jonathan Pritchard (Genetics) having previously worked with Daphne Koller prior to her move to Coursera. I did my PhD with Zoubin Ghahramani in the Machine Learning group of the Cambridge University Engineering Department. I was the Roger Needham Scholar at Wolfson College, funded by Microsoft Research. My undergraduate degree comprised two years of Physics before switching to Engineering to complete an MEng with Zoubin. I took the MSc Bioinformatics and Systems Biology at Imperial College in 2007/8.
Here's a video of me and my friend Johan falling off cliffs on skis in Flaine, France.
Research
My research involves both the development of novel machine learning methods and their application to data analysis problems in genomics, particularly genetic regulation of the transcriptome.
RNA splicing
We developed LeafCutter to identify, quantify and test variable intron splicing events, obviating the need for accurate transcript annotations and circumventing the challenges in estimating relative isoform abundance. An early version of LeafCutter was used in our study linking complex disease and splicing a paper describing the method is under review, and we actively maintain code.
Using allelic specific expression to detect GxE interaction effects
Detecting gene by environment effects on the transcriptome is challenging in observational data. I developed a computational method, EAGLE, which leverages allele-specific expression as a controlled, with-in individual test of the influence of environment factors on different genetic backgrounds. Code is available on github.
Bayesian nonparametric models
Bayesian nonparametric (BNP) models are a category of statistical methods that automatically adapt their complexity to observed data. I have developed BNP methods for hierarchical clustering, heteroskedastic multivariate regression, network data, variable clustering and nonparametric sparse factor analysis (NSFA). In particular, NSFA is able to adaptively choose an appropriate number of factors from data. I used this method to delineate gene co-expression modules from microarray data, and other researchers have subsequently used it in diverse applications including image denoising and EEG analysis.
Variational methods
Variational methods offer a computationally efficient alternative to Markov chain Monte Carlo algorithms for inference in Bayesian probabilistic models. My work allows such methods to be more easily applied to a broader range of probabilistic models. I extended variational message passing to "non-conjugate" (intuitively, less tractable) models, and incorporated this method, Non-conjugate VMP (NCVMP), into the publicly available Infer.NET software package. Later, Tim Salimans and I did early work on using Monte Carlo estimation within variational learning, which is now an active subfield of research under the moniker "Stochastic Variational Inference".
Publications
Please visit my lab website for a more up-to-date list of publications.Working/under submission
- Calderon D, Nguyen ML, Mezger A, Kathiria A, Nguyen V, Lescano N, Wu B, Trombetta J, Ribado JV, Knowles DA, Gao Z, Parent AV, Burt TD, Anderson MS, Criswell LA, Greenleaf WJ, Marson A and Pritchard JK (2018), "Landscape of stimulation-responsive chromatin across diverse human immune cells", bioRxiv.Abstract: The immune system is controlled by a balanced interplay among specialized cell types transitioning between resting and stimulated states. Despite its importance, the regulatory landscape of this system has not yet been fully characterized. To address this gap, we collected ATAC-seq and RNA-seq data under resting and stimulated conditions for 25 immune cell types from peripheral blood of four healthy individuals, and seven cell types from three fetal thymus samples. We found that stimulation caused widespread chromatin remodeling, including a large class of response elements shared between stimulated B and T cells. Furthermore, several autoimmune traits showed significant heritability in stimulation-responsive elements from distinct cell types, highlighting the critical importance of these cell states in autoimmunity. Use of allele-specific read-mapping identified thousands of variants that alter chromatin accessibility in particular conditions. Notably, variants associated with changes in stimulation-specific chromatin accessibility were not enriched for associations with gene expression regulation in whole blood -- a tissue commonly used in eQTL studies. Thus, large-scale maps of variants associated with gene regulation lack a condition important for understanding autoimmunity. As a proof-of-principle we identified variant rs6927172, which links stimulated T cell-specific chromatin dysregulation in the TNFAIP3 locus to ulcerative colitis and rheumatoid arthritis. Overall, our results provide a broad resource of chromatin landscape dynamics and highlight the need for large-scale characterization of effects of genetic variation in stimulated cells.BibTeX:
@article{Calderon2018immune, author = {Calderon, Diego and Nguyen, Michelle L.T. and Mezger, Anja and Kathiria, Arwa and Nguyen, Vinh and Lescano, Ninnia and Wu, Beijing and Trombetta, John and Ribado, Jessica V. and Knowles, David A. and Gao, Ziyue and Parent, Audrey V. and Burt, Trevor D. and Anderson, Mark S. and Criswell, Lindsey A. and Greenleaf, William J. and Marson, Alexander and Pritchard, Jonathan K.}, title = {Landscape of stimulation-responsive chromatin across diverse human immune cells}, journal = {bioRxiv}, publisher = {Cold Spring Harbor Laboratory}, year = {2018}, url = {https://www.biorxiv.org/content/early/2018/09/05/409722}, doi = {10.1101/409722} } - Wainberg M, Sinnott-Armstrong N, Knowles DA, Golan D, Ermel R, Ruusalepp A, Quertermous T, Hao K, Bjorkegren JLM, Rivas MA and Kundaje A (2017), "Vulnerabilities of transcriptome-wide association studies", bioRxiv.Abstract: Transcriptome-wide association studies (TWAS) integrate GWAS and expression quantitative trait locus (eQTL) datasets to discover candidate causal gene-trait associations. We integrate multi-tissue expression panels and summary GWAS for LDL cholesterol and Crohn's disease to show that TWAS are highly vulnerable to discovering non-causal genes, because variants at a single GWAS hit locus are often eQTLs for multiple genes. TWAS exhibit acute instability when the tissue of the expression panel is changed: candidate causal genes that are TWAS hits in one tissue are usually no longer hits in another, due to lack of expression or strong eQTLs, while non-causal genes at the same loci remain. While TWAS is statistically valid when used as a weighted burden test to identify trait-associated loci, it is invalid to interpret TWAS associations as causal genes because the false discovery rate for TWAS causal gene discovery is not only high, but unquantifiable. More broadly, our results showcase limitations of using expression variation across individuals to determine causal genes at GWAS loci.BibTeX:
@article{Wainberg206961, author = {Wainberg, Michael and Sinnott-Armstrong, Nasa and Knowles, David A and Golan, David and Ermel, Raili and Ruusalepp, Arno and Quertermous, Thomas and Hao, Ke and Bjorkegren, Johan L. M. and Rivas, Manuel A. and Kundaje, Anshul}, title = {Vulnerabilities of transcriptome-wide association studies}, journal = {bioRxiv}, publisher = {Cold Spring Harbor Laboratory}, year = {2017}, url = {https://www.biorxiv.org/content/early/2017/10/27/206961}, doi = {10.1101/206961} } - Knowles DA (2015), "Stochastic gradient variational Bayes for gamma approximating distributions", arXiv. , pp. 1509.01631.Abstract: While stochastic variational inference is relatively well known for scaling inference in Bayesian probabilistic models, related methods also offer ways to circumnavigate the approximation of analytically intractable expectations. The key challenge in either setting is controlling the variance of gradient estimates: recent work has shown that for continuous latent variables, particularly multivariate Gaussians, this can be achieved by using the gradient of the log posterior. In this paper we apply the same idea to gamma distributed latent variables given gamma variational distributions, enabling straightforward "black box" variational inference in models where sparsity and non-negativity are appropriate. We demonstrate the method on a recently proposed gamma process model for network data, as well as a novel sparse factor analysis. We outperform generic sampling algorithms and the approach of using Gaussian variational distributions on transformed variables.BibTeX:
@article{Knowles2015stochastic, author = {Knowles, David A}, title = {Stochastic gradient variational Bayes for gamma approximating distributions}, journal = {arXiv}, year = {2015}, pages = {1509.01631}, url = {https://arxiv.org/abs/1509.01631} } - Salimans T and Knowles DA (2014), "On using control variates with stochastic approximation for variational Bayes and its connection to stochastic linear regression", arXiv. , pp. 1401.1022.Abstract: Recently, we and several other authors have written about the possibilities of using stochastic approximation techniques for fitting variational approximations to intractable Bayesian posterior distributions. Naive implementations of stochastic approximation suffer from high variance in this setting. Several authors have therefore suggested using control variates to reduce this variance, while we have taken a different but analogous approach to reducing the variance which we call stochastic linear regression. In this note we take the former perspective and derive the ideal set of control variates for stochastic approximation variational Bayes under a certain set of assumptions. We then show that using these control variates is closely related to using the stochastic linear regression approximation technique we proposed earlier. A simple example shows that our method for constructing control variates leads to stochastic estimators with much lower variance compared to other approaches.BibTeX:
@article{salimans2014using, author = {Salimans, Tim and Knowles, David A}, title = {On using control variates with stochastic approximation for variational Bayes and its connection to stochastic linear regression}, journal = {arXiv}, year = {2014}, pages = {1401.1022}, url = {https://arxiv.org/abs/1401.1022} } - Palla* K, Knowles* DA and Ghahramani Z (2013), "A dependent partition-valued process for multitask clustering and time evolving network modeling", arXiv. , pp. 1303.3265. *These authors contributed equally to this work.Abstract: The fundamental aim of clustering algorithms is to partition data points. We consider tasks where the discovered partition is allowed to vary with some covariate such as space or time. One approach would be to use fragmentation-coagulation processes, but these, being Markov processes, are restricted to linear or tree structured covariate spaces. We define a partition-valued process on an arbitrary covariate space using Gaussian processes. We use the process to construct a multitask clustering model which partitions datapoints in a similar way across multiple data sources, and a time series model of network data which allows cluster assignments to vary over time. We describe sampling algorithms for inference and apply our method to defining cancer subtypes based on different types of cellular characteristics, finding regulatory modules from gene expression data from multiple human populations, and discovering time varying community structure in a social network.BibTeX:
@article{palla2013dependent, author = {Palla*, Konstantina and Knowles*, David A and Ghahramani, Zoubin}, title = {A dependent partition-valued process for multitask clustering and time evolving network modeling}, journal = {arXiv}, year = {2013}, pages = {1303.3265}, url = {https://arxiv.org/abs/1303.3265} }
Genetics
- Knowles* DA, Burrows* CK, Blischak JD, Patterson KM, Serie DJ, Norton N, Ober C, Pritchard JK and Gilad Y (2018), "Determining the genetic basis of anthracycline-cardiotoxicity by molecular response QTL mapping in induced cardiomyocytes", eLife. *These authors contributed equally to this work.Abstract: Anthracycline-induced cardiotoxicity (ACT) is a key limiting factor in setting optimal chemotherapy regimes for cancer patients, with almost half of patients expected to ultimately develop congestive heart failure given high drug doses. However, the genetic basis of sensitivity to anthracyclines such as doxorubicin remains unclear. To begin addressing this, we created a panel of iPSC-derived cardiomyocytes from 45 individuals and performed RNA-seq after 24h exposure to varying levels of doxorubicin. The transcriptomic response to doxorubicin is substantial, with the majority of genes being differentially expressed across treatments of different concentrations and over 6000 genes showing evidence of differential splicing. Overall, our observations indicate that splicing fidelity decreases in the presence of doxorubicin. We detect 376 response-expression QTLs and 42 response-splicing QTLs, i.e. genetic variants that modulate the individual transcriptomic response to doxorubicin in terms of expression and splicing changes respectively. We show that inter-individual variation in transcriptional response is predictive of cell damage measured in vitro using a cardiac troponin assay, which in turn is shown to be associated with in vivo ACT risk. Finally, the molecular QTLs we detected are enriched in lower ACT GWAS p-values, further supporting the in vivo relevance of our map of genetic regulation of cellular response to anthracyclines.BibTeX:
@article{Knowles2018dox, author = {Knowles*, David A and Burrows*, Courtney K and Blischak, John D and Patterson, Kristen M and Serie, Daniel J. and Norton, Nadine and Ober, Carole and Pritchard, Jonathan K and Gilad, Yoav}, title = {Determining the genetic basis of anthracycline-cardiotoxicity by molecular response QTL mapping in induced cardiomyocytes}, journal = {eLife}, year = {2018}, url = {https://elifesciences.org/articles/33480}, doi = {10.7554/eLife.33480} } - Leland Taylor D, Knowles DA, Scott LJ, Ramirez AH, Casale FP, Wolford BN, Guan L, Varshney A, Albanus RD, Parker SCJ, Narisu N, Chines PS, Erdos MR, Welch RP, Kinnunen L, Saramies J, Sundvall J, Lakka TA, Laakso M, Tuomilehto J, Koistinen HA, Stegle O, Boehnke M, Birney E and Collins FS (2018), "Interactions between genetic variation and cellular environment in skeletal muscle gene expression", PLoS One., April, 2018. Vol. 13(4), pp. e0195788.Abstract: From whole organisms to individual cells, responses to environmental conditions are influenced by genetic makeup, where the effect of genetic variation on a trait depends on the environmental context. RNA-sequencing quantifies gene expression as a molecular trait, and is capable of capturing both genetic and environmental effects. In this study, we explore opportunities of using allele-specific expression (ASE) to discover cis-acting genotype-environment interactions (GxE)---genetic effects on gene expression that depend on an environmental condition. Treating 17 common, clinical traits as approximations of the cellular environment of 267 skeletal muscle biopsies, we identify 10 candidate environmental response expression quantitative trait loci (reQTLs) across 6 traits (12 unique gene-environment trait pairs; 10% FDR per trait) including sex, systolic blood pressure, and low-density lipoprotein cholesterol. Although using ASE is in principle a promising approach to detect GxE effects, replication of such signals can be challenging as validation requires harmonization of environmental traits across cohorts and a sufficient sampling of heterozygotes for a transcribed SNP. Comprehensive discovery and replication will require large human transcriptome datasets, or the integration of multiple transcribed SNPs, coupled with standardized clinical phenotyping.BibTeX:
@article{Leland_Taylor2018-lb, author = {Leland Taylor, D and Knowles, David A and Scott, Laura J and Ramirez, Andrea H and Casale, Francesco Paolo and Wolford, Brooke N and Guan, Li and Varshney, Arushi and Albanus, Ricardo D'oliveira and Parker, Stephen C J and Narisu, Narisu and Chines, Peter S and Erdos, Michael R and Welch, Ryan P and Kinnunen, Leena and Saramies, Jouko and Sundvall, Jouko and Lakka, Timo A and Laakso, Markku and Tuomilehto, Jaakko and Koistinen, Heikki A and Stegle, Oliver and Boehnke, Michael and Birney, Ewan and Collins, Francis S}, title = {Interactions between genetic variation and cellular environment in skeletal muscle gene expression}, journal = {PLoS One}, publisher = {Public Library of Science}, year = {2018}, volume = {13}, number = {4}, pages = {e0195788}, doi = {10.1371/journal.pone.0195788} } - Li* YI, Knowles* DA, Humphrey J, Barbeira AN, Dickinson SP, Im HK and Pritchard JK (2017), "Annotation-free quantification of RNA splicing using LeafCutter", Nature Genetics. , pp. 044107. *These authors contributed equally to this work.Abstract: The excision of introns from pre-mRNA is an essential step in mRNA processing. We developed LeafCutter to study sample and population variation in intron splicing. LeafCutter identifies variable intron splicing events from short-read RNA-seq data and finds alternative splicing events of high complexity. Our approach obviates the need for transcript annotations and overcomes the challenges in estimating relative isoform or exon usage in complex splicing events. LeafCutter can be used both for detecting differential splicing between sample groups, and for mapping splicing quantitative trait loci (sQTLs). Compared to contemporary methods, we find over three times more sQTLs, many of which help us ascribe molecular effects to disease-associated variants. LeafCutter is fast, easy to use, and available at https://github.com/davidaknowles/BibTeX:
@article{LeafCutter, author = {Li*, Yang I. and Knowles*, David A. and Humphrey, Jack and Barbeira, Alvaro N. and Dickinson, Scott P. and Im, Hae Kyung and Pritchard, Jonathan K.}, title = {Annotation-free quantification of RNA splicing using LeafCutter}, journal = {Nature Genetics}, year = {2017}, pages = {044107}, url = {https://www.nature.com/articles/s41588-017-0004-9}, doi = {10.1038/s41588-017-0004-9} } - Knowles DA, Davis JR, Edgington H, Raj A, Favé M-J, Zhu X, Potash JB, Weissman MM, Shi J, Levinson D, Awadalla P, Mostafavi S, Montgomery SB and Battle A (2017), "Allele-specific expression reveals interactions between genetic variation and environment", Nature Methods.Abstract: Identifying interactions between genetics and the environment (GxE) remains challenging. We have developed EAGLE, a hierarchical Bayesian model for identifying GxE interactions based on association between environment and allele-specific expression (ASE). Combining RNA-sequencing of whole blood and extensive environmental annotations collected from 922 human individuals, we identified 35 GxE interactions, compared to only four using standard GxE testing. EAGLE provides new opportunities to identify GxE interactions using functional genomic data.BibTeX:
@article{Knowles2017gxe, author = {Knowles, David A and Davis, Joe R and Edgington, Hilary and Raj, Anil and Favé, Marie-Julie and Zhu, Xiaowei and Potash, James B and Weissman, Myrna M and Shi, Jianxin and Levinson, Doug and Awadalla, Philip and Mostafavi, Sara and Montgomery, Stephen B and Battle, Alexis}, title = {Allele-specific expression reveals interactions between genetic variation and environment}, journal = {Nature Methods}, year = {2017}, url = {http://www.nature.com/nmeth/journal/vaop/ncurrent/full/nmeth.4298.html}, doi = {10.1038/nmeth.4298} } - Tung P-Y, Blischak JD, Hsiao CJ, Knowles DA, Burnett JE, Pritchard JK and Gilad Y (2017), "Batch effects and the effective design of single-cell gene expression studies", Scientific Reports. Vol. 7, pp. 39921.Abstract: Single cell RNA sequencing (scRNA-seq) can be used to characterize variation in gene expression levels at high resolution. However, the sources of experimental noise in scRNA-seq are not yet well understood. We investigated the technical variation associated with sample processing using the single cell Fluidigm C1 platform. To do so, we processed three C1 replicates from three human induced pluripotent stem cell (iPSC) lines. We added unique molecular identifiers (UMIs) to all samples, to account for amplification bias. We found that the major source of variation in the gene expression data was driven by genotype, but we also observed substantial variation between the technical replicates. We observed that the conversion of reads to molecules using the UMIs was impacted by both biological and technical variation, indicating that UMI counts are not an unbiased estimator of gene expression levels. Based on our results, we suggest a framework for effective scRNA-seq studies.BibTeX:
@article{Tung2017, author = {Tung, Po-Yuan and Blischak, John D. and Hsiao, Chiaowen Joyce and Knowles, David A. and Burnett, Jonathan E. and Pritchard, Jonathan K. and Gilad, Yoav}, title = {Batch effects and the effective design of single-cell gene expression studies}, journal = {Scientific Reports}, year = {2017}, volume = {7}, pages = {39921}, url = {http://www.nature.com/articles/srep39921}, doi = {10.1038/srep39921} } - Calderon D, Bhaskar A, Knowles DA, Golan D, Raj T, Fu A and Pritchard JK (2017), "Inferring Relevant Cell Types For Complex Traits Using Single-Cell Gene Expression", The American Journal of Human Genetics. , pp. 136283.Abstract: Previous studies have prioritized trait-relevant cell types by looking for an enrichment of GWAS signal within functional regions. However, these studies are limited in cell resolution by the lack of functional annotations from difficult-to-characterize or rare cell populations. Measurement of single-cell gene expression has become a popular method for characterizing novel cell types, and yet, hardly any work exists linking single-cell RNA-seq to phenotypes of interest. To address this deficiency, we present RolyPoly, a regression-based polygenic model that can prioritize trait-relevant cell types and genes from GWAS summary statistics and single-cell RNA-seq. We demonstrate RolyPoly's accuracy through simulation and validate previously known tissue-trait associations. We discover a significant association between microglia and late-onset Alzheimer's disease, and an association between oligodendrocytes and replicating fetal cortical cells with schizophrenia. Additionally, RolyPoly computes a trait-relevance score for each gene which reflects the importance of expression specific to a cell type. We found that differentially expressed genes in the prefrontal cortex of Alzheimer's patients were significantly enriched for highly ranked genes by RolyPoly gene scores. Overall, our method represents a powerful framework for understanding the effect of common variants on cell types contributing to complex traits.BibTeX:
@article{Calderon2017, author = {Calderon, Diego and Bhaskar, Anand and Knowles, David A and Golan, David and Raj, Towfique and Fu, Audrey and Pritchard, Jonathan K}, title = {Inferring Relevant Cell Types For Complex Traits Using Single-Cell Gene Expression}, journal = {The American Journal of Human Genetics}, year = {2017}, pages = {136283}, url = {http://biorxiv.org/content/early/2017/05/10/136283.abstract} } - Tsang EK, Abell NS, Li X, Anaya V, Karczewski KJ, Knowles DA, Sierra RG, Smith KS and Montgomery SB (2017), "Small RNA sequencing in cells and exosomes identifies eQTLs and 14q32 as a region of active export", G3 Genes|Genomes|Genetics. Vol. 7(1), pp. 31-39.Abstract: Exosomes are small extracellular vesicles that carry heterogeneous cargo, including RNA, between cells. Increasing evidence suggests that exosomes are important mediators of intercellular communication and biomarkers of disease. Despite this, the variability of exosomal RNA between individuals has not been well quantified. To assess this variability, we sequenced the small RNA of cells and exosomes from a 17-member family. Across individuals, we show that selective export of miRNAs occurs not only at the level of specific transcripts, but that a cluster of 74 mature miRNAs on chromosome 14q32 is massively exported in exosomes while mostly absent from cells. We also observe more interindividual variability between exosomal samples than between cellular ones and identify four miRNA expression quantitative trait loci shared between cells and exosomes. Our findings indicate that genomically colocated miRNAs can be exported together and highlight the variability in exosomal miRNA levels between individuals as relevant for exosome use as diagnostics.BibTeX:
@article{Tsang2017, author = {Tsang, Emily K. and Abell, Nathan S. and Li, Xin and Anaya, Vanessa and Karczewski, Konrad J. and Knowles, David A. and Sierra, Raymond G. and Smith, Kevin S. and Montgomery, Stephen B.}, title = {Small RNA sequencing in cells and exosomes identifies eQTLs and 14q32 as a region of active export}, journal = {G3 Genes|Genomes|Genetics}, year = {2017}, volume = {7}, number = {1}, pages = {31--39}, url = {http://g3journal.org/lookup/doi/10.1534/g3.116.036137}, doi = {10.1534/g3.116.036137} } - Becker LA, Huang B, Bieri G, Ma R, Knowles DA, Jafar-Nejad P, Messing J, Kim HJ, Soriano A, Auburger G, Pulst SM, Taylor JP, Rigo F and Gitler AD (2017), "Therapeutic reduction of ataxin-2 extends lifespan and reduces pathology in TDP-43 mice", Nature. Vol. 544(7650), pp. 367-371.Abstract: Amyotrophic lateral sclerosis (ALS) is a rapidly progressing neurodegenerative disease that is characterized by motor neuron loss and that leads to paralysis and death 2--5 years after disease onset 1 . Nearly all patients with ALS have aggregates of the RNA-binding protein TDP-43 in their brains and spinal cords 2 , and rare mutations in the gene encoding TDP-43 can cause ALS 3 . There are no effective TDP-43-directed therapies for ALS or related TDP-43 proteinopathies, such as frontotemporal dementia. Antisense oligonucleotides (ASOs) and RNA-interference approaches are emerging as attractive therapeutic strategies in neurological diseases 4 . Indeed, treatment of a rat model of inherited ALS (caused by a mutation in Sod1) with ASOs against Sod1 has been shown to substantially slow disease progression 5 . However, as SOD1 mutations account for only around 2--5% of ALS cases, additional therapeutic strategies are needed. Silencing TDP-43 itself is probably not appropriate, given its critical cellular functions 1,6 . Here we present a promising alternative therapeutic strategy for ALS that involves targeting ataxin-2. A decrease in ataxin-2 suppresses TDP-43 toxicity in yeast and flies 7 , and intermediate-length polyglutamine expansions in the ataxin-2 gene increase risk of ALS 7,8 . We used two independent approaches to test whether decreasing ataxin-2 levels could mitigate disease in a mouse model of TDP-43 proteinopathy 9 . First, we crossed ataxin-2 knockout mice with TDP-43 (also known as TARDBP) transgenic mice. The decrease in ataxin-2 reduced aggregation of TDP-43, markedly increased survival and improved motor function. Second, in a more therapeutically applicable approach, we administered ASOs targeting ataxin-2 to the central nervous system of TDP-43 transgenic mice. This single treatment markedly extended survival. Because TDP-43 aggregation is a component of nearly all cases of ALS 6 , targeting ataxin-2 could represent a broadly effective therapeutic strategy. To test the hypothesis that a decrease in ataxin-2 levels can res-cue neurodegenerative phenotypes caused by TDP-43 accumula-tion, we first used a genetic approach. There are several transgenic mouse lines that express wild-type or mutant TDP-43, using various strategies 10BibTeX:
@article{Becker2017, author = {Becker, Lindsay A. and Huang, Brenda and Bieri, Gregor and Ma, Rosanna and Knowles, David A. and Jafar-Nejad, Paymaan and Messing, James and Kim, Hong Joo and Soriano, Armand and Auburger, Georg and Pulst, Stefan M. and Taylor, J. Paul and Rigo, Frank and Gitler, Aaron D.}, title = {Therapeutic reduction of ataxin-2 extends lifespan and reduces pathology in TDP-43 mice}, journal = {Nature}, year = {2017}, volume = {544}, number = {7650}, pages = {367--371}, url = {http://www.nature.com/doifinder/10.1038/nature22038}, doi = {10.1038/nature22038} } - Davis JR, Fresard L, Knowles DA, Pala M, Bustamante CD, Battle A and Montgomery SB (2016), "An Efficient Multiple-Testing Adjustment for eQTL Studies that Accounts for Linkage Disequilibrium between Variants", The American Journal of Human Genetics. Vol. 98(1), pp. 216-224.Abstract: Methods for multiple-testing correction in local expression quantitative trait locus (cis-eQTL) studies are a trade-off between statistical power and computational efficiency. Bonferroni correction, though computationally trivial, is overly conservative and fails to account for linkage disequilibrium between variants. Permutation-based methods are more powerful, though computationally far more intensive. We present an alternative correction method called eigenMT, which runs over 500 times faster than permutations and has adjusted p values that closely approximate empirical ones. To achieve this speed while also maintaining the accuracy of permutation-based methods, we estimate the effective number of independent variants tested for association with a particular gene, termed Meff, by using the eigenvalue decomposition of the genotype correlation matrix. We employ a regularized estimator of the correlation matrix to ensure Meff is robust and yields adjusted p values that closely approximate p values from permutations. Finally, using a common genotype matrix, we show that eigenMT can be applied with even greater efficiency to studies across tissues or conditions. Our method provides a simpler, more efficient approach to multiple-testing correction than existing methods and fits within existing pipelines for eQTL discovery.BibTeX:
@article{Davis2016eigenmt, author = {Davis, Joe R. and Fresard, Laure and Knowles, David A. and Pala, Mauro and Bustamante, Carlos D. and Battle, Alexis and Montgomery, Stephen B.}, title = {An Efficient Multiple-Testing Adjustment for eQTL Studies that Accounts for Linkage Disequilibrium between Variants}, journal = {The American Journal of Human Genetics}, year = {2016}, volume = {98}, number = {1}, pages = {216--224}, url = {http://www.cell.com/ajhg/abstract/S0002-9297(15)00492-9}, doi = {10.1016/j.ajhg.2015.11.021} } - Kukurba KR, Parsana P, Balliu B, Smith KS, Zappala Z, Knowles DA, Favé M-J, Davis JR, Li X, Zhu X, Potash JB, Weissman MM, Shi J, Kundaje A, Levinson DF, Awadalla P, Mostafavi S, Battle A and Montgomery SB (2016), "Impact of the X chromosome and sex on regulatory variation", Genome Research. Vol. 26(6), pp. 768-777.Abstract: The X chromosome, with its unique mode of inheritance, contributes to differences between the sexes at a molecular level, including sex-specific gene expression and sex-specific impact of genetic variation. We have conducted an analysis of the impact of both sex and the X chromosome on patterns of gene expression identified through transcriptome sequencing of whole blood from 922 individuals. We identified that genes on the X chromosome are more likely to have sex-specific expression compared to the autosomal genes. Furthermore, we identified a depletion of regulatory variants on the X chromosome, especially among genes under high selective constraint. In contrast, we discovered an enrichment of sex-specific regulatory variants on the X chromosome. To resolve the molecular mechanisms underlying such effects, we generated and connected sex-specific chromatin accessibility to sex-specific expression and regulatory variation. As sex-specific regulatory variants can inform sex differences in genetic disease prevalence, we have integrated our data with genome-wide association study data for multiple immune traits and to identify traits with significant sex biases. Together, our study provides genome-wide insight into how the X chromosome and sex shape human gene regulation and disease.BibTeX:
@article{Kukurba2015, author = {Kukurba, Kimberly R. and Parsana, Princy and Balliu, Brunilda and Smith, Kevin S. and Zappala, Zachary and Knowles, David A. and Favé, Marie-Julie and Davis, Joe R. and Li, Xin and Zhu, Xiaowei and Potash, James B. and Weissman, Myrna M. and Shi, Jianxin and Kundaje, Anshul and Levinson, Douglas F. and Awadalla, Philip and Mostafavi, Sara and Battle, Alexis and Montgomery, Stephen B.}, title = {Impact of the X chromosome and sex on regulatory variation}, journal = {Genome Research}, publisher = {Cold Spring Harbor Labs Journals}, year = {2016}, volume = {26}, number = {6}, pages = {768--777}, url = {http://genome.cshlp.org/lookup/doi/10.1101/gr.197897.115}, doi = {10.1101/gr.197897.115} } - Li YI, van de Geijn B, Raj A, Knowles DA, Petti AA, Golan D, Gilad Y and Pritchard JK (2016), "RNA splicing is a primary link between genetic variation and disease.", Science. Vol. 352(6285), pp. 600-4.Abstract: Noncoding variants play a central role in the genetics of complex traits, but we still lack a full understanding of the molecular pathways through which they act. We quantified the contribution of cis-acting genetic effects at all major stages of gene regulation from chromatin to proteins, in Yoruba lymphoblastoid cell lines (LCLs). About 65% of expression quantitative trait loci (eQTLs) have primary effects on chromatin, whereas the remaining eQTLs are enriched in transcribed regions. Using a novel method, we also detected 2893 splicing QTLs, most of which have little or no effect on gene-level expression. These splicing QTLs are major contributors to complex traits, roughly on a par with variants that affect gene expression levels. Our study provides a comprehensive view of the mechanisms linking genetic variation to variation in human gene regulation.BibTeX:
@article{Li2016splicing, author = {Li, Yang I and van de Geijn, Bryce and Raj, Anil and Knowles, David A and Petti, Allegra A and Golan, David and Gilad, Yoav and Pritchard, Jonathan K}, title = {RNA splicing is a primary link between genetic variation and disease.}, journal = {Science}, publisher = {American Association for the Advancement of Science}, year = {2016}, volume = {352}, number = {6285}, pages = {600--4}, url = {http://www.ncbi.nlm.nih.gov/pubmed/27126046}, doi = {10.1126/science.aad9417} } - Kukurba KR, Zhang R, Li X, Smith KS, Knowles DA, How Tan M, Piskol R, Lek M, Snyder M, MacArthur DG, Li JB and Montgomery SB (2014), "Allelic Expression of Deleterious Protein-Coding Variants across Human Tissues", PLoS Genetics. Vol. 10(5), pp. e1004304.Abstract: Personal exome and genome sequencing provides access to loss-of-function and rare deleterious alleles whose interpretation is expected to provide insight into individual disease burden. However, for each allele, accurate interpretation of its effect will depend on both its penetrance and the trait's expressivity. In this regard, an important factor that can modify the effect of a pathogenic coding allele is its level of expression; a factor which itself characteristically changes across tissues. To better inform the degree to which pathogenic alleles can be modified by expression level across multiple tissues, we have conducted exome, RNA and deep, targeted allele-specific expression (ASE) sequencing in ten tissues obtained from a single individual. By combining such data, we report the impact of rare and common loss-of-function variants on allelic expression exposing stronger allelic bias for rare stop-gain variants and informing the extent to which rare deleterious coding alleles are consistently expressed across tissues. This study demonstrates the potential importance of transcriptome data to the interpretation of pathogenic protein-coding variants.BibTeX:
@article{Kukurba2014deleterious, author = {Kukurba, Kimberly R. and Zhang, Rui and Li, Xin and Smith, Kevin S. and Knowles, David A. and How Tan, Meng and Piskol, Robert and Lek, Monkol and Snyder, Michael and MacArthur, Daniel G. and Li, Jin Billy and Montgomery, Stephen B.}, title = {Allelic Expression of Deleterious Protein-Coding Variants across Human Tissues}, journal = {PLoS Genetics}, publisher = {Public Library of Science}, year = {2014}, volume = {10}, number = {5}, pages = {e1004304}, url = {http://dx.plos.org/10.1371/journal.pgen.1004304}, doi = {10.1371/journal.pgen.1004304} } - Li X, Battle A, Karczewski KJ, Zappala Z, Knowles DA, Smith KS, Kukurba KR, Wu E, Simon N and Montgomery SB (2014), "Transcriptome sequencing of a large human family identifies the impact of rare noncoding variants.", American Journal of Human Genetics. Vol. 95(3), pp. 245-56.Abstract: Recent and rapid human population growth has led to an excess of rare genetic variants that are expected to contribute to an individual's genetic burden of disease risk. To date, much of the focus has been on rare protein-coding variants, for which potential impact can be estimated from the genetic code, but determining the impact of rare noncoding variants has been more challenging. To improve our understanding of such variants, we combined high-quality genome sequencing and RNA sequencing data from a 17-individual, three-generation family to contrast expression quantitative trait loci (eQTLs) and splicing quantitative trait loci (sQTLs) within this family to eQTLs and sQTLs within a population sample. Using this design, we found that eQTLs and sQTLs with large effects in the family were enriched with rare regulatory and splicing variants (minor allele frequency 0.01). They were also more likely to influence essential genes and genes involved in complex disease. In addition, we tested the capacity of diverse noncoding annotation to predict the impact of rare noncoding variants. We found that distance to the transcription start site, evolutionary constraint, and epigenetic annotation were considerably more informative for predicting the impact of rare variants than for predicting the impact of common variants. These results highlight that rare noncoding variants are important contributors to individual gene-expression profiles and further demonstrate a significant capability for genomic annotation to predict the impact of rare noncoding variants..BibTeX:
@article{Li2014rare, author = {Li, Xin and Battle, Alexis and Karczewski, Konrad J. and Zappala, Zach and Knowles, David A. and Smith, Kevin S. and Kukurba, Kim R. and Wu, Eric and Simon, Noah and Montgomery, Stephen B.}, title = {Transcriptome sequencing of a large human family identifies the impact of rare noncoding variants.}, journal = {American Journal of Human Genetics}, publisher = {Elsevier}, year = {2014}, volume = {95}, number = {3}, pages = {245--56}, url = {http://www.cell.com/article/S0002929714003486/fulltext}, doi = {10.1016/j.ajhg.2014.08.004} } - Glass D, Viñuela A, Davies MN, Ramasamy A, Parts L, Knowles DA, Brown AA, Hedman AK, Small KS, Buil A, Grundberg E, Nica AC, Meglio P, Nestle FO, Ryten M, Durbin R, McCarthy MI, Deloukas P, Dermitzakis ET, Weale ME, Bataille V and Spector TD (2013), "Gene expression changes with age in skin, adipose tissue, blood and brain.", Genome biology. Vol. 14(7), pp. R75.Abstract: BACKGROUND: Previous studies have demonstrated that gene expression levels change with age. These changes are hypothesized to influence the aging rate of an individual. We analyzed gene expression changes with age in abdominal skin, subcutaneous adipose tissue and lymphoblastoid cell lines in 856 female twins in the age range of 39-85 years. Additionally, we investigated genotypic variants involved in genotype-by-age interactions to understand how the genomic regulation of gene expression alters with age. RESULTS: Using a linear mixed model, differential expression with age was identified in 1,672 genes in skin and 188 genes in adipose tissue. Only two genes expressed in lymphoblastoid cell lines showed significant changes with age. Genes significantly regulated by age were compared with expression profiles in 10 brain regions from 100 postmortem brains aged 16 to 83 years. We identified only one age-related gene common to the three tissues. There were 12 genes that showed differential expression with age in both skin and brain tissue and three common to adipose and brain tissues. CONCLUSIONS: Skin showed the most age-related gene expression changes of all the tissues investigated, with many of the genes being previously implicated in fatty acid metabolism, mitochondrial activity, cancer and splicing. A significant proportion of age-related changes in gene expression appear to be tissue-specific with only a few genes sharing an age effect in expression across tissues. More research is needed to improve our understanding of the genetic influences on aging and the relationship with age-related diseases.BibTeX:
@article{Glass2013muther, author = {Glass, Daniel and Viñuela, Ana and Davies, Matthew N and Ramasamy, Adaikalavan and Parts, Leopold and Knowles, David A. and Brown, Andrew A and Hedman, Asa K and Small, Kerrin S and Buil, Alfonso and Grundberg, Elin and Nica, Alexandra C and Meglio, Paoladi and Nestle, Frank O and Ryten, Mina and Durbin, Richard and McCarthy, Mark I and Deloukas, Panagiotis and Dermitzakis, Emmanouil T and Weale, Michael E and Bataille, Veronique and Spector, Tim D}, title = {Gene expression changes with age in skin, adipose tissue, blood and brain.}, journal = {Genome biology}, year = {2013}, volume = {14}, number = {7}, pages = {R75}, url = {http://genomebiology.com/2013/14/7/R75}, doi = {10.1186/gb-2013-14-7-r75} } - Grundberg E, Small KS, Hedman AK, Nica AC, Buil A, Keildson S, Bell JT, Yang T-P, Meduri E, Barrett A, Nisbett J, Sekowska M, Wilk A, Shin S-Y, Glass D, Travers M, Min JL, Knowles DA, Ring S, Ho K, Thorleifsson G, Kong A, Thorsteindottir U, Ainali C, Dimas AS, Hassanali N, Ingle C, Krestyaninova M, Lowe CE, Di Meglio P, Montgomery SB, Parts L, Potter S, Surdulescu G, Tsaprouni L, Tsoka S, Bataille V, Durbin R, Nestle FO, O'Rahilly S, Soranzo N, Lindgren CM, Zondervan KT, Ahmadi KR, Schadt EE, Stefansson K, Smith GD, McCarthy MI, Deloukas P, Dermitzakis ET and Spector TD (2012), "Mapping cis- and trans-regulatory effects across multiple tissues in twins.", Nature Genetics. Vol. 44(10), pp. 1084-9.Abstract: Sequence-based variation in gene expression is a key driver of disease risk. Common variants regulating expression in cis have been mapped in many expression quantitative trait locus (eQTL) studies, typically in single tissues from unrelated individuals. Here, we present a comprehensive analysis of gene expression across multiple tissues conducted in a large set of mono- and dizygotic twins that allows systematic dissection of genetic (cis and trans) and non-genetic effects on gene expression. Using identity-by-descent estimates, we show that at least 40% of the total heritable cis effect on expression cannot be accounted for by common cis variants, a finding that reveals the contribution of low-frequency and rare regulatory variants with respect to both transcriptional regulation and complex trait susceptibility. We show that a substantial proportion of gene expression heritability is trans to the structural gene, and we identify several replicating trans variants that act predominantly in a tissue-restricted manner and may regulate the transcription of many genes.BibTeX:
@article{Grundberg2012, author = {Grundberg, Elin and Small, Kerrin S and Hedman, Asa K and Nica, Alexandra C and Buil, Alfonso and Keildson, Sarah and Bell, Jordana T and Yang, Tsun-Po and Meduri, Eshwar and Barrett, Amy and Nisbett, James and Sekowska, Magdalena and Wilk, Alicja and Shin, So-Youn and Glass, Daniel and Travers, Mary and Min, Josine L and Knowles, David A. and Ring, Sue and Ho, Karen and Thorleifsson, Gudmar and Kong, Augustine and Thorsteindottir, Unnur and Ainali, Chrysanthi and Dimas, Antigone S and Hassanali, Neelam and Ingle, Catherine and Krestyaninova, Maria and Lowe, Christopher E and Di Meglio, Paola and Montgomery, Stephen B and Parts, Leopold and Potter, Simon and Surdulescu, Gabriela and Tsaprouni, Loukia and Tsoka, Sophia and Bataille, Veronique and Durbin, Richard and Nestle, Frank O and O'Rahilly, Stephen and Soranzo, Nicole and Lindgren, Cecilia M and Zondervan, Krina T and Ahmadi, Kourosh R and Schadt, Eric E and Stefansson, Kari and Smith, George Davey and McCarthy, Mark I and Deloukas, Panos and Dermitzakis, Emmanouil T and Spector, Tim D.}, title = {Mapping cis- and trans-regulatory effects across multiple tissues in twins.}, journal = {Nature Genetics}, year = {2012}, volume = {44}, number = {10}, pages = {1084--9}, url = {http://dx.doi.org/10.1038/ng.2394}, doi = {10.1038/ng.2394} } - Schöne C, Venner A, Knowles DA, Karnani MM and Burdakov D (2011), "Dichotomous cellular properties of mouse orexin/hypocretin neurons.", The Journal of Physiology. Vol. 589(Pt 11), pp. 2767-79.Abstract: Hypothalamic hypocretin/orexin (Hcrt/Orx) neurons recently emerged as critical regulators of sleep--wake cycles, reward seeking and body energy balance. However, at the level of cellular and network properties, it remains unclear whether Hcrt/Orx neurons are one homogeneous population, or whether there are several distinct types of Hcrt/Orx cells. Here, we collated diverse structural and functional information about individual Hcrt/Orx neurons in mouse brain slices, by combining patch-clamp analysis of spike firing, membrane currents and synaptic inputs with confocal imaging of cell shape and subsequent 3-dimensional Sholl analysis of dendritic architecture. Statistical cluster analysis of intrinsic firing properties revealed that Hcrt/Orx neurons fall into two distinct types. These two cell types also differ in the complexity of their dendritic arbour, the strength of AMPA and GABAA receptor-mediated synaptic drive that they receive, and the density of low-threshold, 4-aminopyridine-sensitive, transient K+ current. Our results provide quantitative evidence that, at the cellular level, the mouse Hcrt/Orx system is composed of two classes of neurons with different firing properties, morphologies and synaptic input organization.BibTeX:
@article{Schone2011, author = {Schöne, Cornelia and Venner, Anne and Knowles, David A. and Karnani, Mahesh M and Burdakov, Denis}, title = {Dichotomous cellular properties of mouse orexin/hypocretin neurons.}, journal = {The Journal of Physiology}, year = {2011}, volume = {589}, number = {Pt 11}, pages = {2767--79}, url = {http://jp.physoc.org/content/early/2011/04/11/jphysiol.2011.208637.abstract}, doi = {10.1113/jphysiol.2011.208637} } - Movassagh M, Choy M-K, Knowles DA, Cordeddu L, Haider S, Down T, Siggens L, Vujic A, Simeoni I, Penkett C, Goddard M, Lio P, Bennett M and Foo R (2011), "Distinct Epigenomic Features in End-Stage Failing Human Hearts", Circulation, American Heart Association. Vol. 135Abstract: BACKGROUND:
The epigenome refers to marks on the genome, including DNA methylation and histone modifications, that regulate the expression of underlying genes. A consistent profile of gene expression changes in end-stage cardiomyopathy led us to hypothesize that distinct global patterns of the epigenome may also exist.METHODS AND RESULTS:
We constructed genome-wide maps of DNA methylation and histone-3 lysine-36 trimethylation (H3K36me3) enrichment for cardiomyopathic and normal human hearts. More than 506 Mb sequences per library were generated by high-throughput sequencing, allowing us to assign methylation scores to ≈28 million CG dinucleotides in the human genome. DNA methylation was significantly different in promoter CpG islands, intragenic CpG islands, gene bodies, and H3K36me3-enriched regions of the genome. DNA methylation differences were present in promoters of upregulated genes but not downregulated genes. H3K36me3 enrichment itself was also significantly different in coding regions of the genome. Specifically, abundance of RNA transcripts encoded by the DUX4 locus correlated to differential DNA methylation and H3K36me3 enrichment. In vitro, Dux gene expression was responsive to a specific inhibitor of DNA methyltransferase, and Dux siRNA knockdown led to reduced cell viability.CONCLUSIONS:
Distinct epigenomic patterns exist in important DNA elements of the cardiac genome in human end-stage cardiomyopathy. The epigenome may control the expression of local or distal genes with critical functions in myocardial stress response. If epigenomic patterns track with disease progression, assays for the epigenome may be useful for assessing prognosis in heart failure. Further studies are needed to determine whether and how the epigenome contributes to the development of cardiomyopathy.BibTeX:@article{Movassagh2011a, author = {Movassagh, Mehregan and Choy, Mun-Kit and Knowles, David A and Cordeddu, Lina and Haider, Syed and Down, Thomas and Siggens, Lee and Vujic, Ana and Simeoni, Ilenia and Penkett, Chris and Goddard, Martin and Lio, Pietro and Bennett, Martin and Foo, Roger}, title = {Distinct Epigenomic Features in End-Stage Failing Human Hearts}, journal = {Circulation, American Heart Association}, year = {2011}, volume = {135}, url = {http://circ.ahajournals.org/content/early/2011/10/24/CIRCULATIONAHA.111.040071.abstract}, doi = {10.1161/CIRCULATIONAHA.111.040071} } - Glass D, Parts L, Knowles D, Aviv A and Spector TD (2010), "No correlation between childhood maltreatment and telomere length.", Biological psychiatry. Vol. 68(6), pp. e21-2.BibTeX:
@article{Glass2010, author = {Glass, Daniel and Parts, Leopold and Knowles, David and Aviv, Abraham and Spector, Tim D}, title = {No correlation between childhood maltreatment and telomere length.}, journal = {Biological psychiatry}, year = {2010}, volume = {68}, number = {6}, pages = {e21--2}, url = {http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2930212/}, doi = {10.1016/j.biopsych.2010.02.026} } - Knowles DA and Holmes S (2009), "Statistical tools for ultra-deep pyrosequencing of fast evolving viruses", NIPS Workshop: Computational Biology. , pp. 1-9.Abstract: We aim to detect minor variant Hepatitis B viruses (HBV) in 38 pyrosequencing samples from infected individuals. Errors involved in the amplification and ultra deep pyrosequencing (UDPS) of these samples are characterised using HBV plasmid controls. Homopolymeric regions and quality scores are found to be significant covariates in determining insertion and deletion (indel) error rates, but not mismatch rates which depend on the nucleotide transition matrix. This knowledge is used to derive two methods for classifying genuine mutations: a hypothesis testing framework and a mixture model. Using an approximate "ground truth" from a limiting dilution Sanger sequencing run, these methods are shown to outperform the naive percentage threshold approach. The possibility of early stage PCR errors becoming significant is investigated by simulation, which underlines the importance of the initial copy number.BibTeX:
@article{Knowles2009pyroseq, author = {Knowles, David A. and Holmes, Susan}, title = {Statistical tools for ultra-deep pyrosequencing of fast evolving viruses}, journal = {NIPS Workshop: Computational Biology}, year = {2009}, pages = {1--9}, url = {http://mlg.eng.cam.ac.uk/pub/pdf/KnoHol09.pdf} }
Machine learning/statistics
- Palla* K, Knowles* DA and Ghahramani Z (2017), "A birth-death process for feature allocation.", In Proceedings of the 34th International Conference on Machine Learning. *These authors contributed equally to this work.Abstract: We propose a Bayesian nonparametric prior over feature allocations for sequential data, the birth-death feature allocation process (BDFP). The BDFP models the evolution of the feature allocation of a set of N objects across a covariate (e.g.time) by creating and deleting features. A BDFP is exchangeable, projective, stationary and reversible, and its equilibrium distribution is given by the Indian buffet process (IBP). We also show that the Beta process on an extended space is the de Finetti mixing distribution underlying the BDFP. Finally, we present the finite approximation of the BDFP, the Beta Event Process (BEP), that permits simplified inference. The utility of the BDFP as a prior is demonstrated on real world dynamic genomics and social network data.BibTeX:
@inproceedings{palla2017bdfp, author = {Palla*, Konstantina and Knowles*, David A. and Ghahramani, Zoubin}, title = {A birth-death process for feature allocation.}, booktitle = {Proceedings of the 34th International Conference on Machine Learning}, year = {2017} } - Shah A, Knowles D and Ghahramani Z (2015), "An Empirical Study of Stochastic Variational Inference Algorithms for the Beta Bernoulli Process", In Proceedings of the 32nd International Conference on Machine Learning. , pp. 1594-1603.Abstract: Stochastic variational inference (SVI) is emerging as the most promising candidate for scaling inference in Bayesian probabilistic models to large datasets. However, the performance of these methods has been assessed primarily in the context of Bayesian topic models, particularly latent Dirichlet allocation (LDA). Deriving several new algorithms, and using synthetic, image and genomic datasets, we investigate whether the understanding gleaned from LDA applies in the setting of sparse latent factor models, specifically beta process factor analysis (BPFA). We demonstrate that the big picture is consistent: using Gibbs sampling within SVI to maintain certain posterior dependencies is extremely effective. However, we find that different posterior dependencies are important in BPFA relative to LDA. Particularly, approximations able to model intra-local variable dependence perform best.BibTeX:
@inproceedings{Shah2015, author = {Shah, Amar and Knowles, David and Ghahramani, Zoubin}, title = {An Empirical Study of Stochastic Variational Inference Algorithms for the Beta Bernoulli Process}, booktitle = {Proceedings of the 32nd International Conference on Machine Learning}, year = {2015}, pages = {1594--1603}, url = {http://proceedings.mlr.press/v37/shahb15.pdf} } - Knowles DA and Ghahramani Z (2015), "Pitman Yor Diffusion Trees for Bayesian hierarchical clustering", IEEE Transactions on Pattern Analysis and Machine Intelligence. Vol. 37(2), pp. 271-289.Abstract: In this paper we introduce the Pitman Yor Diffusion Tree (PYDT), a Bayesian non-parametric prior over tree structures which generalises the Dirichlet Diffusion Tree [Neal, 2001] and removes the restriction to binary branching structure. The generative process is described and shown to result in an exchangeable distribution over data points. We prove some theoretical properties of the model including showing its construction as the continuum limit of a nested Chinese restaurant process model. We then present two alternative MCMC samplers which allows us to model uncertainty over tree structures, and a computationally efficient greedy Bayesian EM search algorithm. Both algorithms use message passing on the tree structure. The utility of the model and algorithms is demonstrated on synthetic and real world data, both continuous and binary.BibTeX:
@article{Knowles2014, author = {Knowles, David A. and Ghahramani, Zoubin}, title = {Pitman Yor Diffusion Trees for Bayesian hierarchical clustering}, journal = {IEEE Transactions on Pattern Analysis and Machine Intelligence}, publisher = {IEEE Computer Society}, year = {2015}, volume = {37}, number = {2}, pages = {271--289}, url = {https://dx.doi.org/10.1109/TPAMI.2014.2313115}, doi = {10.1109/TPAMI.2014.2313115} } - Palla K, Knowles DA and Ghahramani Z (2015), "Relational learning and network modelling using infinite latent attribute models", IEEE Transactions on Pattern Analysis and Machine Intelligence Special Issue on Bayesian Nonparametrics. Vol. 37(2), pp. 462-474.Abstract: Latent variable models for network data extract a summary of the relational structure underlying an observed network. The simplest possible models subdivide nodes of the network into clusters; the probability of a link between any two nodes then depends only on their cluster assignment. Currently available models can be classified by whether clusters are disjoint or are allowed to overlap. These models can explain a flat clustering structure. Hierarchical Bayesian models provide a natural approach to capture more complex dependencies. We propose a model in which objects are characterised by a latent feature vector. Each feature is itself partitioned into disjoint groups (subclusters), corresponding to a second layer of hierarchy. In experimental comparisons, the model achieves significantly improved predictive performance on social and biological link prediction tasks. The results indicate that models with a single layer hierarchy over-simplify real networks.BibTeX:
@article{Palla2015, author = {Palla, Konstantina and Knowles, David A. and Ghahramani, Zoubin}, title = {Relational learning and network modelling using infinite latent attribute models}, journal = {IEEE Transactions on Pattern Analysis and Machine Intelligence Special Issue on Bayesian Nonparametrics}, year = {2015}, volume = {37}, number = {2}, pages = {462--474}, doi = {10.1109/TPAMI.2014.2324586} } - Knowles DA, Palla K and Ghahramani Z (2014), "A reversible infinite HMM using normalised random measures", In Proceedings of The 31st International Conference on Machine Learning.Abstract: We present a nonparametric prior over reversible Markov chains. We use completely random measures, specifically gamma processes, to construct a countably infinite graph with weighted edges. By enforcing symmetry to make the edges undirected we define a prior over random walks on graphs that results in a reversible Markov chain. The resulting prior over infinite transition matrices is closely related to the hierarchical Dirichlet process but enforces reversibility. A reinforcement scheme has recently been proposed with similar properties, but the de Finetti measure is not well characterised. We take the alternative approach of explicitly constructing the mixing measure, which allows more straightforward and efficient inference at the cost of no longer having a closed form predictive distribution. We use our process to construct a reversible infinite HMM which we apply to two real datasets, one from epigenomics and one ion channel recording.BibTeX:
@inproceedings{Knowles2014a, author = {Knowles, David A. and Palla, Konstantina and Ghahramani, Zoubin}, title = {A reversible infinite HMM using normalised random measures}, booktitle = {Proceedings of The 31st International Conference on Machine Learning}, year = {2014}, url = {http://proceedings.mlr.press/v32/knowles14.pdf} } - Heaukulani C, Knowles DA and Ghahramani Z (2014), "Beta Diffusion Trees", In Proceedings of the 31st International Conference on Machine Learning. , pp. 1809-1817.Abstract: We define the beta diffusion tree, a random tree structure with a set of leaves that defines a collection of overlapping subsets of objects, known as a feature allocation. A generative process for the tree structure is defined in terms of particles (representing the objects) diffusing in some continuous space, analogously to the Dirichlet diffusion tree (Neal, 2003b), which defines a tree structure over partitions (i.e., non-overlapping subsets) of the objects. Unlike in the Dirichlet diffusion tree, multiple copies of a particle may exist and diffuse along multiple branches in the beta diffusion tree, and an object may therefore belong to multiple subsets of particles. We demonstrate how to build a hierarchically-clustered factor analysis model with the beta diffusion tree and how to perform inference over the random tree structures with a Markov chain Monte Carlo algorithm. We conclude with several numerical experiments on missing data problems with data sets of gene expression microarrays, international development statistics, and intranational socioeconomic measurements.BibTeX:
@inproceedings{Heaukulani2014beta, author = {Heaukulani, Creighton and Knowles, David A. and Ghahramani, Zoubin}, title = {Beta Diffusion Trees}, booktitle = {Proceedings of the 31st International Conference on Machine Learning}, year = {2014}, pages = {1809--1817}, url = {http://proceedings.mlr.press/v32/heaukulani14.pdf} } - Salimans T and Knowles DA (2013), "Fixed-form variational posterior approximation through stochastic linear regression", Bayesian Analysis. Vol. 8(4), pp. 837-882. Winner of the International Society for Bayesian Analysis Lindley Prize..Abstract: We propose a general algorithm for approximating nonstandard Bayesian posterior distributions. The algorithm minimizes the Kullback-Leibler divergence of an approximating distribution to the intractable posterior distribution. Our method can be used to approximate any posterior distribution, provided that it is given in closed form up to the proportionality constant. The approximation can be any distribution in the exponential family or any mixture of such distributions, which means that it can be made arbitrarily precise. Several examples illustrate the speed and accuracy of our approximation method in practice.BibTeX:
@article{salimans2013, author = {Salimans, Tim and Knowles, David A.}, title = {Fixed-form variational posterior approximation through stochastic linear regression}, journal = {Bayesian Analysis}, publisher = {International Society for Bayesian Analysis}, year = {2013}, volume = {8}, number = {4}, pages = {837--882}, url = {http://projecteuclid.org/euclid.ba/1386166315}, doi = {10.1214/13-BA858} } - Quadrianto N, Sharmanska V, Knowles DA and Ghahramani Z (2013), "The Supervised IBP: Neighbourhood Preserving Infinite Latent Feature Models", In Proceedings of the 29th Conference on Uncertainty in Artificial Intelligence.Abstract: We propose a probabilistic model to infer supervised latent variables in the Hamming space from observed data. Our model allows simultaneous inference of the number of binary latent variables, and their values. The latent variables preserve neighbourhood structure of the data in a sense that objects in the same semantic concept have similar latent values, and objects in different concepts have dissimilar latent values. We formulate the supervised infinite latent variable problem based on an intuitive principle of pulling objects together if they are of the same type, and pushing them apart if they are not. We then combine this principle with a flexible Indian Buffet Process prior on the latent variables. We show that the inferred supervised latent variables can be directly used to perform a nearest neighbour search for the purpose of retrieval. We introduce a new application of dynamically extending hash codes, and show how to effectively couple the structure of the hash codes with continuously growing structure of the neighbourhood preserving infinite latent feature space.BibTeX:
@inproceedings{quadrianto2013supervised, author = {Quadrianto, Novi and Sharmanska, Viktoriia and Knowles, David A and Ghahramani, Zoubin}, title = {The Supervised IBP: Neighbourhood Preserving Infinite Latent Feature Models}, booktitle = {Proceedings of the 29th Conference on Uncertainty in Artificial Intelligence}, year = {2013}, url = {http://mlg.eng.cam.ac.uk/pub/pdf/QuaShaKnoGha13.pdf} } - Palla* K, Knowles* DA and Ghahramani Z (2012), "A nonparametric variable clustering model", In Advances in Neural Information Processing Systems. Vol. 5, pp. 2987-2995. *These authors contributed equally to this work.Abstract: Factor analysis models effectively summarise the covariance structure of high dimensional data, but the solutions are typically hard to interpret. This motivates attempting to find a disjoint partition, i.e. a clustering, of observed variables so that variables in a cluster are highly correlated. We introduce a Bayesian non-parametric approach to this problem, and demonstrate advantages over heuristic methods proposed to date.BibTeX:
@inproceedings{Palla2012nonparametric, author = {Palla*, Konstantina and Knowles*, David A. and Ghahramani, Zoubin}, title = {A nonparametric variable clustering model}, booktitle = {Advances in Neural Information Processing Systems}, year = {2012}, volume = {5}, pages = {2987--2995}, url = {https://papers.nips.cc/paper/4579-a-nonparametric-variable-clustering-model} } - Palla* K, Knowles* DA and Ghahramani Z (2012), "An Infinite Latent Attribute Model for Network Data", In Proceedings of the 29th International Conference on Machine Learning. , pp. 1607-1614. *These authors contributed equally to this work.Abstract: Latent variable models for network data extract a summary of the relational structure underlying an observed network. The simplest possible models subdivide nodes of the network into clusters; the probability of a link between any two nodes then depends only on their cluster assignment. Currently available models can be classified by whether clusters are disjoint or are allowed to overlap. These models can explain clustering structure. Hierarchical Bayesian models provide a natural approach to capture more complex dependencies. We propose a model in which objects are characterised by a latent feature vector. Each feature is itself partitioned into disjoint groups (subclusters), corresponding to a second layer of hierarchy. In experimental comparisons, the model achieves significantly improved predictive performance on social and biological link prediction tasks. The results indicate that models with a single layer hierarchy over-simplify real networks.BibTeX:
@inproceedings{palla2012infinite, author = {Palla*, Konstantina and Knowles*, David A. and Ghahramani, Zoubin}, title = {An Infinite Latent Attribute Model for Network Data}, booktitle = {Proceedings of the 29th International Conference on Machine Learning}, year = {2012}, pages = {1607--1614}, url = {http://icml.cc/2012/papers/785.pdf} } - Wilson AG, Knowles DA and Ghahramani Z (2012), "Gaussian Process Regression Networks", In Proceedings of the 29th International Conference on Machine Learning. , pp. 599-606.Abstract: We introduce a new regression frame- work, Gaussian process regression networks (GPRN), which combines the structural properties of Bayesian neural networks with the nonparametric flexibility of Gaussian pro- cesses. GPRN accommodates input (pre- dictor) dependent signal and noise corre- lations between multiple output (response) variables, input dependent length-scales and amplitudes, and heavy-tailed predictive dis- tributions. We derive both elliptical slice sampling and variational Bayes inference pro- cedures for GPRN. We apply GPRN as a multiple output regression and multivariate volatility model, demonstrating substantially improved performance over eight popular multiple output (multi-task) Gaussian pro- cess models and three multivariate volatility models on real datasets, including a 1000 di- mensional gene expression dataset.BibTeX:
@inproceedings{wilson2011gaussian, author = {Wilson, Andrew Gordon and Knowles, David A. and Ghahramani, Zoubin}, title = {Gaussian Process Regression Networks}, booktitle = {Proceedings of the 29th International Conference on Machine Learning}, year = {2012}, pages = {599--606}, url = {http://icml.cc/2012/papers/329.pdf} } - Knowles DA, Gael JV and Ghahramani Z (2011), "Message Passing Algorithms for the Dirichlet Diffusion Tree", In Proceedings of the 28th International Conference on Machine Learning. , pp. 721-728.Abstract: We demonstrate efficient approximate inference for the Dirichlet Diffusion Tree (Neal, 2003), a Bayesian nonparametric prior over tree structures. Although DDTs provide a powerful and elegant approach for modeling hierarchies they haven't seen much use to date. One problem is the computational cost of MCMC inference. We provide the first deterministic approximate inference methods for DDT models and show excellent performance compared to the MCMC alternative. We present message passing algorithms to approximate the Bayesian model evidence for a specific tree. This is used to drive sequential tree building and greedy search to find optimal tree structures, corresponding to hierarchical clusterings of the data. We demonstrate appropriate observation models for continuous and binary data. The empirical performance of our method is very close to the computationally expensive MCMC alternative on a density estimation problem, and significantly outperforms kernel density estimators.BibTeX:
@inproceedings{Gael2011, author = {Knowles, David A. and Gael, Jurgen Van and Ghahramani, Zoubin}, title = {Message Passing Algorithms for the Dirichlet Diffusion Tree}, booktitle = {Proceedings of the 28th International Conference on Machine Learning}, year = {2011}, pages = {721--728}, url = {http://www.icml-2011.org/papers/410_icmlpaper.pdf} } - Knowles DA and Minka T (2011), "Non-conjugate Variational Message Passing for Multinomial and Binary Regression", In Advances in Neural Information Processing Systems. , pp. 1701-1709.Abstract: Variational Message Passing (VMP) is an algorithmic implementation of the Variational Bayes (VB) method which applies only in the special case of conjugate exponential family models. We propose an extension to VMP, which we refer to as Non-conjugate Variational Message Passing (NCVMP) which aims to alleviate this restriction while maintaining modularity, allowing choice in how expectations are calculated, and integrating into an existing message-passing framework: Infer.NET. We demonstrate NCVMP on logistic binary and multinomial regression. In the multinomial case we introduce a novel variational bound for the softmax factor which is tighter than other commonly used bounds whilst maintaining computational tractability.BibTeX:
@inproceedings{Knowles2011c, author = {Knowles, David A. and Minka, Tom}, title = {Non-conjugate Variational Message Passing for Multinomial and Binary Regression}, booktitle = {Advances in Neural Information Processing Systems}, year = {2011}, pages = {1701--1709}, url = {http://papers.nips.cc/paper/4407-non-conjugate-variational-message-passing-for-multinomial-and-binary-regression} } - Knowles DA and Ghahramani Z (2011), "Nonparametric Bayesian sparse factor models with application to gene expression modeling", The Annals of Applied Statistics. Vol. 5(2B), pp. 1534-1552.Abstract: A nonparametric Bayesian extension of Factor Analysis (FA) is proposed where observed data Y is modeled as a linear superposition, G, of a potentially infinite number of hidden factors, X. The Indian Buffet Process (IBP) is used as a prior on G to incorporate sparsity and to allow the number of latent features to be inferred. The model's utility for modeling gene expression data is investigated using randomly generated datasets based on a known sparse connectivity matrix for E. Coli, and on three biological datasets of increasing complexity.BibTeX:
@article{Knowles2011nonparametric, author = {Knowles, David A. and Ghahramani, Zoubin}, title = {Nonparametric Bayesian sparse factor models with application to gene expression modeling}, journal = {The Annals of Applied Statistics}, publisher = {Institute of Mathematical Statistics}, year = {2011}, volume = {5}, number = {2B}, pages = {1534--1552}, url = {https://projecteuclid.org/euclid.aoas/1310562732}, doi = {10.1214/10-AOAS435} } - Knowles DA and Ghahramani Z (2011), "Pitman-Yor Diffusion Trees", In Proceedings of the 27th Conference on Uncertainty in Artificial Intelligence. , pp. 410-418.Abstract: We introduce the Pitman Yor Diffusion Tree (PYDT) for hierarchical clustering, a generalization of the Dirichlet Diffusion Tree (Neal, 2001) which removes the restriction to binary branching structure. The generative process is described and shown to result in an exchangeable distribution over data points. We prove some theoretical properties of the model and then present two inference methods: a collapsed MCMC sampler which allows us to model uncertainty over tree structures, and a computationally efficient greedy Bayesian EM search algorithm. Both algorithms use message passing on the tree structure. The utility of the model and algorithms is demonstrated on synthetic and real world data, both continuous and binary.BibTeX:
@inproceedings{Knowles2011b, author = {Knowles, David A. and Ghahramani, Zoubin}, title = {Pitman-Yor Diffusion Trees}, booktitle = {Proceedings of the 27th Conference on Uncertainty in Artificial Intelligence}, year = {2011}, pages = {410--418}, url = {http://dl.acm.org/citation.cfm?id=3020596} } - Doshi-Velez* F, Mohamed* S, Knowles* DA and Ghahramani Z (2009), "Large Scale Nonparametric Bayesian Inference: Data Parallelisation in the Indian Buffet Process", In Advances in Neural Information Processing Systems. , pp. 1294-1302. *These authors contributed equally to this work.Abstract: Nonparametric Bayesian models provide a framework for flexible probabilistic modelling of complex datasets. Unfortunately, Bayesian inference methods often require high-dimensional averages and can be slow to compute, especially with the potentially unbounded representations associated with nonparametric models. We address the challenge of scaling nonparametric Bayesian inference to the increasingly large datasets found in real-world applications, focusing on the case of parallelising inference in the Indian Buffet Process (IBP). Our approach divides a large data set between multiple processors. The processors use message passing to compute likelihoods in an asynchronous, distributed fashion and to propagate statistics about the global Bayesian posterior. This novel MCMC sampler is the first parallel inference scheme for IBP-based models, scaling to datasets orders of magnitude larger than had previously been possible.BibTeX:
@inproceedings{Doshi-velez2009, author = {Doshi-Velez*, Finale and Mohamed*, Shakir and Knowles*, David A. and Ghahramani, Zoubin}, title = {Large Scale Nonparametric Bayesian Inference: Data Parallelisation in the Indian Buffet Process}, booktitle = {Advances in Neural Information Processing Systems}, year = {2009}, pages = {1294--1302}, url = {http://papers.nips.cc/paper/3669-large-scale-nonparametric-bayesian-inference-data-parallelisation-in-the-indian-buffet-process} } - Knowles DA and Ghahramani Z (2007), "Infinite Sparse Factor Analysis and Infinite Independent Components Analysis", In 7th International Conference on Independent Component Analysis and Signal Separation.Abstract: A nonparametric Bayesian extension of Independent Components Analysis (ICA) is proposed where observed data Y is modelled as a linear superposition, G, of a potentially infinite number of hidden sources, X. Whether a given source is active for a specific data point is specified by an infinite binary matrix, Z. The resulting sparse representation allows increased data reduction compared to standard ICA. We define a prior on Z using the Indian Buffet Process (IBP). We describe four variants of the model, with Gaussian or Laplacian priors on X and the one or two-parameter IBPs. We demonstrate Bayesian inference under these models using a Markov Chain Monte Carlo (MCMC) algorithm on synthetic and gene expression data and compare to standard ICA algorithms.BibTeX:
@inproceedings{Knowles07iica, author = {Knowles, David A. and Ghahramani, Zoubin}, title = {Infinite Sparse Factor Analysis and Infinite Independent Components Analysis}, booktitle = {7th International Conference on Independent Component Analysis and Signal Separation}, year = {2007}, url = {http://www.springerlink.com/index/10.1007/978-3-540-74494-8}, doi = {10.1007/978-3-540-74494-8} }
Software
Most of my code is on github, in particular:- LeafCutter RNA splicing tool.
- Detecting gene x environment interaction effects using allele-specific expression..
- C#/Infer.NET code for Gaussian Process Regression Networks is on github.
- Matlab code for nonparametric sparse factor analysis is available here.
- The MCMC sampler for Dirichlet Process Variable Clustering is at https://github.com/davidaknowles/dpvc
Reports/Abstracts/Presentations
Workshop papers/conference abstracts
- Yang Li, Bryce van de Geijn, Allegra Petti, Anil Raj, David A. Knowles, John Blischak, Yoav Gilad, Jonathan Pritchard (2015).
The effects of human genetic variation on the gene regulatory cascade.
American Society of Human Genetics 65th Annual Meeting - David A. Knowles, Stanley Ho, Kien Nguyen, Don Morris, Anthony Magliocco, Anindya Sarkar, Daphne Koller, Sylvia Plevritis, Srinivas Chukka, Michael Barnes (2015).
Machine Learning-based Prognostication of Breast Cancer Recurrence using Tissue Slide Features.
Pathology Visions Winner: Best Poster in Image Analysis!
- David A. Knowles, Joe R. Davis, Stephen B. Montgomery, Alexis Battle (2015).
Detecting gene-by-environment interactions using allele specific expression.
The Biology of Genomes Meeting (CSHL)
- David A. Knowles, Stanley Ho, Kien Nguyen, Don Morris, Anthony Magliocco, Anindya Sarkar, Daphne Koller, Srinivas Chukka, Michael Barnes (2014)
Machine learning-based prognostication of breast cancer recurrence using tissue slide features from H&E and immunohistochemically stained slides.
San Antonio Breast Cancer Symposium
- Emily K. Tsang, Xin Li, Vanessa Anaya, Konrad J. Karczewski, David A. Knowles, Kevin S. Smith, Stepehn B. Montgomery (2014).
Dissecting the genetic regulation of exosome RNA cargo in a large family.
American Society of Human Genetics 64th Annual Meeting - J.R. Davis, D.A. Knowles, S.B. Montgomery, A. Battle (2014)
Rare variation and the genomic context of allele-specific expression. American Society of Human Genetics 64th Annual Meeting
- David A. Knowles, Alexis Battle, Daphne Koller (2013)
Discovering latent cancer characteristics predictive of drug sensitivity.
RECOMB/ISCB Conference on Regulatory & Systems Genomics (selected for oral presentation) - Alexis Battle*, David A. Knowles*, Sara Mostafavi, Xiaowei Zhu, James B. Potash, Myrna M. Weissman, Courtney McCormick, Christian D. Haudenschild, Kenneth B. Beckman, Jianxin Shi, Rui Mei, Alexander E. Urban, Douglas F. Levinson, Daphne Koller, Stephen B. Montgomery (2013)
The relationship between common environmental and genetic effects on human gene splicing and expression.
American Society of Human Genetics (ASHG) Annual Meeting - David A. Knowles, Leopold Parts, Daniel Glass and John M. Winn
Inferring a measure of physiological age from multiple ageing related phenotypes. paper video
To appear at the NIPS workshop: From Statistical Genetics to Predictive Models in Personalized Medicine (NIPS PM 2011) - David A. Knowles, Leopold Parts, Daniel Glass and John M. Winn (2010)
Modeling skin and ageing phenotypes using latent variable models in Infer.NET. paper poster
Poster presented at: Predictive Models in Personalized Medicine Workshop, NIPS 2010, 6-11 December 2010, Vancouver, BC, Canada. - Knowles, D. and Holmes, S. (2009)
Statistical tools for ultra-deep pyrosequencing of fast evolving viruses. pdf video slides
Presented at: Computational Biology Workshop, NIPS 2009, 7-12 December 2009, Vancouver, BC, Canada.
Reports/Theses
- Bayesian non-parametric models and inference for sparse and hierarchical latent structure (2012) pdf
PhD Thesis, University of Cambridge
Supervisor: Zoubin Ghahramani - Serial and Parallel Inference in Sparse Nonparametric Latent Factor Models applied to Gene Expression Modeling (2009) pdf
PhD First Year Report, Department of Engineering, University of Cambridge
Supervisor: Zoubin Ghahramani - Statistical tools for ulta-deep pyrosequencing of fast evolving viruses (2008) pdf
MSc Bioinformatics and Systems Biology, Imperial College London, Individual Project
Supervisor: Professor Susan Holmes, Stanford University - SBML-ABC: a package for data simulation, parameter inference and model selection, Group Report (2008) pdf
MSc Bioinformatics and Systems Biology, Imperial College London, Group Project
Supervisor: Professor Michael Stumpf - Infinite Independent Components Analysis (2007) pdf
MEng Information Engineering, Cambridge University, 4th year project
Supervisor: Professor Zoubin Ghahramani - Real Time Continuous Curvature Path Planner for an Autonomous Vehicle in an Urban Environment (2006) pdf
Summer Undergraduate Research Fellowship, Caltech. I was a member of Team Caltech, an entry into the DARPA Urban Challenge
Supervisor: Professor Richard Murray
Presentations
- Detecting gene-by-environment interactions using allele specific expression. The Biology of Genomes 2015 (image credit @AlexCagan)
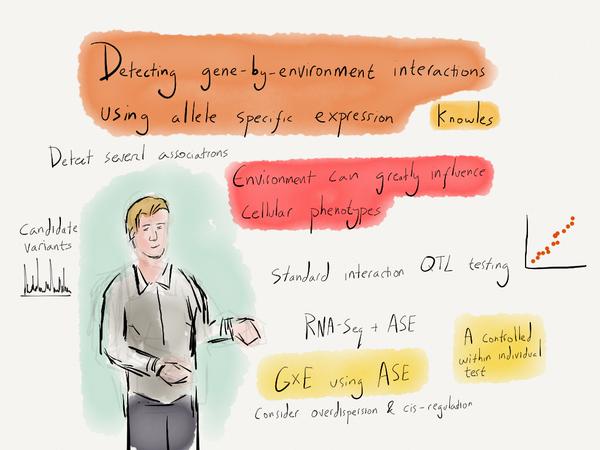
- Properties of Bayesian nonparametric models and priors over trees. Guest lecture as part of Matt Hoffman's STAT300 class, summer 2013.
- Diffusion trees as priors. This was a talk I gave about the Dirichlet diffusion tree and Pitman Yor diffusion tree at Collegio Carlo Alberto.
- Inferring an individual's "physiological" age from multiple ageing-related phenotypes
I gave a talk at the Cambridge Statistics Initiative Special One-Day Meeting, which you can watch here. I also presented this work at the NIPS 2011 Personalised Medicine workshop: paper video - Variational methods for nonparametric Bayesian models
I gave a brief presentation at Microsoft Research summarising some attempts to use variational inference in nonparametric, particularly Dirichlet Process based, models. The slides are here.
Miscellaneous
I was involved with running the Cambridge University Statistics Clinic. At Stanford I help out with SMACC: Statistical, Mathematical, and Computational Consulting.
Lagrangian duality
After presenting at journal club on it, I think I finally got my head round Lagrangian duality, and hopefully came up with a reasonably intuitive explanation. My focus is on intuition rather than rigor, and is based almost entirely on Boyd and Vandenberghe's tome. I thought I should write this up while it's still fresh, so here you go: Lagrangian Duality for Dummies
Binomial p-values
Following my work on 454 pyrosequencing error rates with Professor Holmes, I was asked about how to calculate a p-value for comparing two draws from a Binomial distribution to test the hypothesis that the number of substitutions seen in the sample is significantly greater than the number of substitutions seen in the control. There is actually no need to use the Poisson approximation, and the Binomial distribution very naturally takes care of varying coverage. I explain my approach here.
Infer.NET
During my PhD I was a part-time developer of Infer.NET. I wrote a blog post about some of the features we added in Infer.NET 2.4, see here.